éĆąį╗»į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄūóāį╝╝ąg(sh©┤)īÅ▓ķųĖī¦(d©Żo)įŁät(4)
1.3║¾╠Ä└ĒĘĮĘ©ęį╝░“×ūC
║¾╠Ä└Ē┐╔─▄░³└©ÖC╝ė╣żĪó¤ßĄ╚ņoē║Īó¤ß╠Ä└ĒĪóų¦ō╬╬’╗“Üł┴¶Ę█─®╚ź│²Īó▒Ē├µ╠Ä└ĒĄ╚ĪŻæ¬(y©®ng)«ö(d©Īng)įu╣└║¾╠Ä└Ē╣ż╦ćī”▓─┴Ž║═ĮK«a(ch©Żn)ŲĘĄ─░▓╚½Īóėąą¦ąįĄ─ė░ĒæĪŻ
1.4«a(ch©Żn)ŲĘĄ─£yįć
éĆąį╗»į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄ░ļ│╔ŲĘ║═│╔ŲĘæ¬(y©®ng)«ö(d©Īng)┐╝æ]Ž┬┴ą£yįćŻ║
1.4.1«a(ch©Żn)ŲĘ▓─┴ŽĄ─╗»īW(xu©”)│╔Ęų║═┴”īW(xu©”)ąį─▄æ¬(y©®ng)«ö(d©Īng)Ę¹║Ž╔Ļł¾▓─┴ŽĄ─ŽÓĻP(gu©Īn)ś╦(bi©Īo)£╩(zh©│n)Ż¼└²╚ńā╚(n©©i)▓┐┘|(zh©¼)┴┐Īó’@╬óĮM┐ŚĪó┴”īW(xu©”)ÅŖČ╚ĪóęÄ(gu©®)Č©ĘŪ▒╚└²čė╔ņ┬╩Ą╚ĪŻ
1.4.2«a(ch©Żn)ŲĘ▒Ē├µ┘|(zh©¼)┴┐Īó│▀┤ń╝░Š½Č╚ĪŻįuār«a(ch©Żn)ŲĘ┼c╠ß╣®Ą─3D┤“ėĪĄ─╣Ū„└─Żą═Ą─Ųź┼õąį╝░▀mė├ąįĪŻ
1.4.3«a(ch©Żn)ŲĘā╚(n©©i)▓┐ĮY(ji©”)śŗ(g©░u)Ż¼└²╚ńĘ┬╔·ČÓ┐ūĮY(ji©”)śŗ(g©░u)Ą─┐ūÅĮĪóĮzÅĮĪó┐ūŽČ┬╩Ą╚ĪŻ
1.4.4«a(ch©Żn)ŲĘĄ─╣”─▄ąįįuārŻ¼└²╚ń«a(ch©Żn)ŲĘĄ─ņoæB(t©żi)▌SŽ“ē║┐säéČ╚ĪóņoæB(t©żi)▌SŽ“ē║┐sūŅ┤¾▌d║╔ĪóņoæB(t©żi)▌SŽ“╝¶ŪąūŅ┤¾▌d║╔ĪóäėæB(t©żi)▌SŽ“ē║┐sÅŖČ╚ĪóäėæB(t©żi)▌SŽ“╝¶ŪąÅŖČ╚ĪóņoæB(t©żi)┼ż▐D(zhu©Żn)ūŅ┤¾┼żŠžĪóäėæB(t©żi)┼ż▐D(zhu©Żn)ąį─▄įuārĪóņoæB(t©żi)▌SŽ“ē║┐s│┴Ž▌äéČ╚ĪóäėæB(t©żi)ŲŻä┌Ą╚Ż¼▀@ą®Ęų╬÷æ¬(y©®ng)«ö(d©Īng)┼c«a(ch©Żn)ŲĘŅA(y©┤)Ų┌╩╣ė├▓┐╬╗║═ŅA(y©┤)Ų┌ė├═ŠŽÓ▀mę╦ĪŻ
ūŅ▓ŅŪķørĄ─▀xō±æ¬(y©®ng)«ö(d©Īng)ĮY(ji©”)║Ž«a(ch©Żn)ŲĘ▓─┴Žī┘ąį£yįć║═ėąŽ▐į¬─ŻöMĄ╚╔·╬’┴”īW(xu©”)Ęų╬÷ĪŻ╚ń▒žę¬ĢrŻ¼┐╔═©▀^ī”┐╣ē║─▄┴”Īó┐╣└Ł─▄┴”Īó┐╣┼ż▐D(zhu©Żn)─▄┴”Īó┐╣é╚(c©©)ÅØ─▄┴”Ą─£yįćŻ¼½@Ą├ėąŽ▐į¬Ęų╬÷╦∙ąĶĄ─▓─┴Žī┘ąįģóöĄ(sh©┤)ĪŻ
1.4.5«a(ch©Żn)ŲĘĄ─╔·╬’ŽÓ╚▌ąį£yįćĪŻ
1.4.6«a(ch©Żn)ŲĘĄ─ŪÕŽ┤╝░¤oŠ·Öz£yĪŻ
2.į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄßt(y©®)╣żĮ╗╗ź─▄┴”┤_šJ(r©©n)
2.1éĆąį╗»įO(sh©©)ėŗ
▒Š▓┐Ęųā╚(n©©i)╚▌╩Ūį┌ĪČßt(y©®)»¤Ų„ąĄ╔·«a(ch©Żn)┘|(zh©¼)┴┐╣▄└ĒęÄ(gu©®)ĘČĪĘŻ©ć°╝ę╩│ŲĘ╦ÄŲĘ▒O(ji©Īn)ČĮ╣▄└Ē┐éŠų╣½Ėµ2014─ĻĄ┌64╠¢Ż®įO(sh©©)ėŗ┼cķ_░l(f©Ī)š┬╣Ø(ji©”)Ą─╗∙ĄA(ch©│)╔ŽŻ¼ĮY(ji©”)║ŽéĆąį╗»į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄĄ─╠ž╩ŌąįųŲČ©Ż¼ŅA(y©┤)Ų┌ØMūŃė├ė┌ūóāį╔Ļł¾Ą─éĆąį╗»į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄįO(sh©©)ėŗķ_░l(f©Ī)Ą─╗∙▒Šę¬Ū¾ĪŻ
æ¬(y©®ng)«ö(d©Īng)ė╔┼R┤▓ßt(y©®)╔·Īóė░Ž±┐Ųßt(y©®)╔·Īó╣ż│╠ĤĄ╚ČÓīW(xu©”)┐Ų▒│Š░╚╦åT╣▓═¼ĮMĮ©ßt(y©®)╣żĮ╗╗źłFĻĀĪŻģó┼cßt(y©®)╣żĮ╗╗źįO(sh©©)ėŗĄ─╚╦åTŻ¼æ¬(y©®ng)«ö(d©Īng)Įø(j©®ng)▀^┼cŲõŹÅ╬╗ę¬Ū¾ŽÓ▀mæ¬(y©®ng)Ą─┼Óė¢(x©┤n)Ż¼Š▀ėąŽÓæ¬(y©®ng)└Ēšōų¬ūR║═īŹļH▓┘ū„─▄┴”Ż¼▓óųŲČ©╚╦åT╔ŽŹÅŪ░ßt(y©®)╣żĮ╗╗ź─▄┴”┤_šJ(r©©n)Ą─ś╦(bi©Īo)£╩(zh©│n)▓┘ū„┴„│╠Ż¼├„┤_ŽÓĻP(gu©Īn)╚╦åTį┌įO(sh©©)ėŗķ_░l(f©Ī)ųąĄ─┬Üž¤(z©”)┼cÖÓ(qu©ón)Ž▐ĪŻįO(sh©©)ėŗ┼cķ_░l(f©Ī)Łh(hu©ón)╣Ø(ji©”)Ą─ŽÓĻP(gu©Īn)▀^│╠æ¬(y©®ng)«ö(d©Īng)╬─╝■╗»ĪŻ
2.1.1įO(sh©©)ėŗķ_░l(f©Ī)Ą─▌ö╚ļ
ßt(y©®)╣żĮ╗╗źįO(sh©©)ėŗ╚╦åT╣▓═¼═Ļ│╔«a(ch©Żn)ŲĘįO(sh©©)ėŗŻ¼║×ūų┤_šJ(r©©n)éĆąį╗»į÷▓─ųŲįņ«a(ch©Żn)ŲĘįO(sh©©)ėŗ╦∙ąĶĄ─įO(sh©©)ėŗę¬Ū¾ŪÕå╬Ż¼░³└©╗╝š▀ė░Ž±öĄ(sh©┤)ō■(j©┤)Īó╩ųąg(sh©┤)ĘĮ░ĖĪóéĆąį╗»į÷▓─ųŲįņ«a(ch©Żn)ŲĘę¬Ū¾Ż©▓─┴ŽĪóĮY(ji©”)śŗ(g©░u)Īó│▀┤ńĪó░³čb╝░Į╗ĖČĘĮ╩ĮĄ╚Ż®Īó┼õ╠ū╩╣ė├Ą─Ų„ąĄę¬Ū¾Ą╚ØMūŃ«a(ch©Żn)ŲĘŅA(y©┤)Ų┌ė├═ŠĄ─ŽÓĻP(gu©Īn)ģóöĄ(sh©┤)║═ę¬Ū¾ĪŻ
╗╝š▀ė░Ž±öĄ(sh©┤)ō■(j©┤)æ¬(y©®ng)«ö(d©Īng)░³└©ØMūŃ┼R┤▓ąĶŪ¾Ą─ĻP(gu©Īn)µIģóöĄ(sh©┤)Ż¼ėøõø╦∙╩╣ė├▄ø╝■├¹ĘQ║═░µ▒Š╠¢Ż¼▓╔╚Ī├„┤_┤ļ╩®£p╔┘CT║═MRIÖz▓ķųą║¼Įī┘╝┘¾wĄ─é╬ė░Ż¼▓ó├„┤_┐╔ūĘ╦▌ąįĪŻ▓╔╝»╗╝š▀ė░Ž±öĄ(sh©┤)ō■(j©┤)ĢrŻ¼ę¬┤_▒Żą┼Žó░▓╚½Īó═Ļš¹Ż¼▓ó▓╔╚Ī┐╔┐┐┤ļ╩®▒Żūo╗╝š▀ļ[╦ĮĪŻ╔Ž╩÷ā╚(n©©i)╚▌ė╔ė░Ž±┐Ųßt(y©®)╔·║×ūų┤_šJ(r©©n)ĪŻ
2.1.2įO(sh©©)ėŗķ_░l(f©Ī)“×ūC║═┤_šJ(r©©n)
ßśī”éĆąį╗»į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄĄ─öĄ(sh©┤)ūų╗»─Żą═║═ųŲįņ╝ė╣żŲĘ▀MąąįO(sh©©)ėŗķ_░l(f©Ī)Ą─“×ūC║═┤_šJ(r©©n)ĪŻ┐╔ęį▓╔ė├ę╗ĘN╗“ČÓĘNĘĮĘ©“×ūC«a(ch©Żn)ŲĘī”ĮŌŲ╩Ųź┼õąįĪó╔·╬’┴”īW(xu©”)ąį─▄Ą╚įO(sh©©)ėŗķ_░l(f©Ī)▌ö╚ļ║═ŅA(y©┤)Ų┌ė├═ŠĄ─ØMūŃąįĪŻĘĮĘ©┐╔ęį░³└©ėŗ╦ŃÖC─ŻöMĘų╬÷ĪóīŹ“×╩ęÖz£yĪó┼R┤▓įu╣└Ą╚ĪŻ
«ö(d©Īng)╗╝š▀Ą─öĄ(sh©┤)ō■(j©┤)į┌įŁ“×ūC─Żą═ģóöĄ(sh©┤)ĘČć·ų«ā╚(n©©i)Ż¼┐╔ęį▓╔╚ĪėąŽ▐į¬Ęų╬÷Ą╚įuārĘĮĘ©įu╣└Ųõ’L(f©źng)ļUĪŻ╚ń╣¹╗╝š▀Ą─ĮŌŲ╩║═▓ĪūāĘČć·│¼▀^įŁįO(sh©©)ėŗę¬Ū¾Ż¼æ¬(y©®ng)«ö(d©Īng)ųžą┬▀Mąąįu╣└║═“×ūCĪŻī”ė┌│¼│÷ęč┼·£╩(zh©│n)ūóāįĘČć·Ą─╠žš„ĮY(ji©”)śŗ(g©░u)╝░ģóöĄ(sh©┤)æ¬(y©®ng)«ö(d©Īng)┴Ēąąūóāį╔Ļł¾ĪŻ
įO(sh©©)ėŗ“×ūC║═┤_šJ(r©©n)ā╚(n©©i)╚▌æ¬(y©®ng)«ö(d©Īng)ą╬│╔ĪČéĆąį╗»į÷▓─ųŲįņ«a(ch©Żn)ŲĘįO(sh©©)ėŗĘĮ░ĖĪĘŻ¼ų┴╔┘░³║¼įO(sh©©)ėŗ┴„│╠łDĪó▓─┴Žę¬Ū¾ĪóĮY(ji©”)śŗ(g©░u)╠žš„Īó░³čbĘĮ╩ĮĪóĮ╗ĖČĘĮ╩Į║═ĢrķgĪó«a(ch©Żn)ŲĘ╝╝ąg(sh©┤)ę¬Ū¾Ą╚ā╚(n©©i)╚▌╝░ėøõøĪŻßt(y©®)╣żĮ╗╗źłFĻĀ╣▓═¼┤_šJ(r©©n)▓ó║×ūųĪŻ
2.1.3įO(sh©©)ėŗķ_░l(f©Ī)Ą─Ė³Ė─
į┌éĆąį╗»į÷▓─ųŲįņ«a(ch©Żn)ŲĘĄ─įO(sh©©)ėŗ╗“╔·«a(ch©Żn)▀^│╠ųąŻ¼æ¬(y©®ng)«ö(d©Īng)│õĘų┐╝æ]╗╝š▀▓ĪŪķūā╗»Ą╚ę“╦žī¦(d©Żo)ų┬įO(sh©©)ėŗ▓╗ØMūŃ▌ö╚ļĄ─ŪķørĪŻ╚ń╣¹▀MąąįO(sh©©)ėŗĖ³Ė─Ż¼æ¬(y©®ng)«ö(d©Īng)╠ß╣®│õĘųĄ─└Ēė╔Ż¼į┘┤╬ė╔ßt(y©®)╣żĮ╗╗źłFĻĀ║×ūų┤_šJ(r©©n)ĪŻ
2.2«a(ch©Żn)ŲĘĄ─Į╗ĖČ
«ö(d©Īng)éĆąį╗»ßt(y©®)»¤Ų„ąĄ«a(ch©Żn)ŲĘųŲįņ═Ļ│╔║¾Ż¼į┌Į╗ĖČĮo┼R┤▓ßt(y©®)╔·Ģræ¬(y©®ng)«ö(d©Īng)║×ūų┤_šJ(r©©n)▓ó┤µÖnĪŻ┤µÖnā╚(n©©i)╚▌░³└©éĆąį╗»ßt(y©®)»¤Ų„ąĄĄ─öĄ(sh©┤)ūų╗»─Żą═Īó«a(ch©Żn)ŲĘŠÄ╠¢Īó╗╝š▀ś╦(bi©Īo)ūRĄ╚ĪŻ
2.3«a(ch©Żn)ŲĘĄ─╩╣ė├
Å─╩┬éĆąį╗»į÷▓─ųŲįņßt(y©®)»¤Ų„ąĄ«a(ch©Żn)ŲĘĄ─╔Ļšł╚╦┼cßt(y©®)»¤ÖCśŗ(g©░u)æ¬(y©®ng)«ö(d©Īng)ųŲČ©ŽÓæ¬(y©®ng)Ą─ųŲČ╚Ż¼▓ó╣▓═¼ū±╩žŻ║
2.3.1╩╣ė├éĆąį╗»ßt(y©®)»¤Ų„ąĄķ_š╣╩ųąg(sh©┤)Ą─ßt(y©®)»¤ÖCśŗ(g©░u)æ¬(y©®ng)«ö(d©Īng)Š▀ėąŽÓæ¬(y©®ng)┘Y┘|(zh©¼)Ż¼▒žĒÜį┌ąl(w©©i)╔·ų„╣▄▓┐ķTšJ(r©©n)Č©Ą─Š▀ėąīŻśI(y©©)╝╝ąg(sh©┤)┘YĖ±Ą─ßt(y©®)»¤ÖCśŗ(g©░u)╩╣ė├ĪŻ┼R┤▓ßt(y©®)╔·ų┴╔┘æ¬(y©®ng)«ö(d©Īng)Š▀ėąÅ─śI(y©©)Įø(j©®ng)“ׯ¼▓óĮø(j©®ng)▀^▒žę¬Ą─┼Óė¢(x©┤n)ĪŻ
2.3.2éĆąį╗»ßt(y©®)»¤Ų„ąĄāHė├ė┌ąĶę¬╩╣ė├éĆąį╗»ßt(y©®)»¤Ų„ąĄĄ─╗╝š▀Ż¼╗╝š▀╗“š▀Ųõ▒O(ji©Īn)ūo╚╦æ¬(y©®ng)«ö(d©Īng)║×╩ų¬Ūķ═¼ęŌĢ°ĪŻ╔Ļšł╚╦╝░ßt(y©®)»¤ÖCśŗ(g©░u)ėąÖÓ(qu©ón)½@Ą├╗╝š▀ŽÓæ¬(y©®ng)Ą─öĄ(sh©┤)ō■(j©┤)ą┼ŽóŻ¼═¼ĢrĒÜ▒ŻūCŽÓĻP(gu©Īn)ą┼Žó░▓╚½ĪŻ
2.3.3éĆąį╗»ßt(y©®)»¤Ų„ąĄ╩Ū╗∙ė┌╗╝š▀Ą─ė░Ž±öĄ(sh©┤)ō■(j©┤)▀MąąčąųŲŻ¼┼R┤▓ßt(y©®)╔·æ¬(y©®ng)«ö(d©Īng)▒ŻūC╗╝š▀╚½▓┐įŁ╩╝öĄ(sh©┤)ō■(j©┤)Ą─šµīŹąįĪó£╩(zh©│n)┤_ąį║═┐╔ė├ąįĪŻ
2.3.4┼R┤▓ßt(y©®)╔·æ¬(y©®ng)«ö(d©Īng)ģó┼cš¹¾wĘĮ░ĖĄ─įO(sh©©)ėŗŻ¼▓óī”ūŅĮK«a(ch©Żn)ŲĘĪó┼õ╠ū╩ųąg(sh©┤)╣żŠ▀╝░ŽÓĻP(gu©Īn)╩ųąg(sh©┤)ĘĮ░Ė▀Mąą┤_šJ(r©©n)ĪŻ
2.3.5╬┤╩╣ė├Ą─éĆąį╗»ßt(y©®)»¤Ų„ąĄė╔╔Ļšł╚╦žō(f©┤)ž¤(z©”)╩š╗žŻ¼▓╗Ą├į┘ė├ė┌┼R┤▓ĪŻ
2.3.6╔Ļšł╚╦║═ßt(y©®)»¤ÖCśŗ(g©░u)æ¬(y©®ng)«ö(d©Īng)░┤ššĪČßt(y©®)»¤Ų„ąĄ▓╗┴╝╩┬╝■▒O(ji©Īn)£y║═į┘įuār╣▄└Ē▐kĘ©ĪĘėąĻP(gu©Īn)ęÄ(gu©®)Č©ķ_š╣éĆąį╗»ßt(y©®)»¤Ų„ąĄ▓╗┴╝╩┬╝■▒O(ji©Īn)£y╣żū„ĪŻ
į┌«a(ch©Żn)ŲĘ╚½╔·├³ų▄Ų┌ųąŻ¼╔Ļšł╚╦▀Ćæ¬(y©®ng)«ö(d©Īng)═Ļ│╔ęįŽ┬ā╚(n©©i)╚▌Ż║
2.3.7╔Ļšł╚╦æ¬(y©®ng)«ö(d©Īng)Į©┴óöĄ(sh©┤)ō■(j©┤)ÄņŻ¼ė├ė┌▒Ż┤µ▓Ī╗╝Ą─öĄ(sh©┤)ō■(j©┤)ą┼ŽóŻ¼▓óė╔īŻ╚╦žō(f©┤)ž¤(z©”)ŠSūo▒Ż╣▄ĪŻ│²ĘŪĄ├ĄĮ╗╝š▀╝░ßt(y©®)»¤ÖCśŗ(g©░u)Ą─įS┐╔Ż¼╔Ļšł╚╦▓╗Ą├īóöĄ(sh©┤)ō■(j©┤)╠ß╣®ĮoŲõ╦¹ÖCśŗ(g©░u)╗“éĆ╚╦ĪŻ
2.3.8╔Ļšł╚╦æ¬(y©®ng)«ö(d©Īng)Į©┴ó┐žųŲ│╠ą“Ż¼Č©Ų┌╩š╝»Īóįu╣└éĆąį╗»ßt(y©®)»¤Ų„ąĄ┼R┤▓╩╣ė├ą¦╣¹Ż¼ė├ė┌Ė─▀M«a(ch©Żn)ŲĘąį─▄║═ĮĄĄ═«a(ch©Żn)ŲĘ’L(f©źng)ļUĪŻæ¬(y©®ng)«ö(d©Īng)Į©┴óéĆąį╗»ßt(y©®)»¤Ų„ąĄĄ─╩╣ė├ł¾ĖµųŲČ╚Īóą┼ŽóūĘ╦▌ųŲČ╚Īóį┘įuārųŲČ╚║═ĮKų╣«a(ch©Żn)ŲĘæ¬(y©®ng)ė├ųŲČ╚ĪŻ
2.3.9įuārć└(y©ón)ųž▓╗┴╝╩┬╝■┐╔ęį▓╔ė├░┤ššéĆąį╗»ßt(y©®)»¤Ų„ąĄ╔·«a(ch©Żn)╣ż╦ć╬─╝■Ż¼į┌═¼Ą╚╔·«a(ch©Żn)╝ė╣żŚl╝■Ž┬╔·«a(ch©Żn)Ą─éĆąį╗»ßt(y©®)»¤Ų„ąĄśėŲĘĪŻ╔Ļšł╚╦æ¬(y©®ng)«ö(d©Īng)▒Ż┤µ├┐éĆéĆąį╗»ßt(y©®)»¤Ų„ąĄĄ─įO(sh©©)ėŗ╔·«a(ch©Żn)┘Y┴ŽŻ¼┤_▒Ż├┐éĆéĆąį╗»į÷▓─ųŲįņ«a(ch©Żn)ŲĘĄ─ųž¼F(xi©żn)ąįĪŻ
(ž¤(z©”)╚╬ŠÄ▌ŗŻ║admin)

Ž┬ę╗Ų¬Ż║╚½Ū“╣ż│╠ś╦(bi©Īo)£╩(zh©│n)ģf(xi©”)Ģ■ęč░l(f©Ī)▓╝┴╦╩ūéĆė├ė┌║Į┐š║Į╠ņŅI(l©½ng)ė“Ą─į÷▓─ųŲįņęÄ(gu©®)ĘČ
 MGAßt(y©®)»¤┬ō(li©ón)├╦Ż║ęįŽĄĮy(t©»ng)╗»╦╝
MGAßt(y©®)»¤┬ō(li©ón)├╦Ż║ęįŽĄĮy(t©»ng)╗»╦╝ ć°╝ęĮy(t©»ng)ėŗŠųŻ║ųąć°3D┤“ėĪįO(sh©©)
ć°╝ęĮy(t©»ng)ėŗŠųŻ║ųąć°3D┤“ėĪįO(sh©©) ICCīóųŲČ©╗ņ─²═┴3D┤“ėĪē”
ICCīóųŲČ©╗ņ─²═┴3D┤“ėĪē” ╚²ĒŚį÷▓─ųŲįņĮī┘Ę█─®ć°╝ę
╚²ĒŚį÷▓─ųŲįņĮī┘Ę█─®ć°╝ę ╣żą┼▓┐Ą╚ą┬ęÄ(gu©®)Ż║╣żśI(y©©)╝ē3D┤“
╣żą┼▓┐Ą╚ą┬ęÄ(gu©®)Ż║╣żśI(y©©)╝ē3D┤“ 2027─Ļ«a(ch©Żn)ųĄ═╗ŲŲ1500ā|į¬Ż©
2027─Ļ«a(ch©Żn)ųĄ═╗ŲŲ1500ā|į¬Ż©
- ĪżMGAßt(y©®)»¤┬ō(li©ón)├╦Ż║ęįŽĄĮy(t©»ng)╗»╦╝ŠSŲŲ│²3D┤“ėĪ
- Īżę╗čįŠ┼Č”ļ[ą■ÖCŻ¼┼¬╝┘│╔šµ╚²░╦┌A╩Ū╩▓├┤
- ĪżĮ±Ų┌╦─┴∙║ŽöĄ(sh©┤)ķ_Ż¼╦─┴∙Ž▓ĘĻžö╔±ĄĮ╩Ū╩▓├┤
- Īżę░═Ōßץ─Ż¼ĖŃéƶ~£½╩Ū╩▓├┤╔·ążŻ¼│╔šZßī
- Īż▓╗äė┬Ģ╔½╩Ū╩▓├┤╔·ążŻ¼│╔šZßī┴xĮŌßī┬õīŹ
- Īż╗┼└’╗┼Åł╩Ū╩▓├┤╔·ążŻ¼│╔šZßī┴xĮŌßī┬õīŹ
- ĪżÕe┼õ┘Yį┤Ż¼├„ųķ░Ą═ČŻ¼├„ų¬¤o╣¹æ¬(y©®ng)╩š╩ų╩Ū
- Īż─”ĄŪ╠½╣½Ż¼¤oD¤oŃ^Ż¼║Ą╠’┤╣ß×║╬╦∙Ū¾╩Ū
- ĪżMGAßt(y©®)»¤┬ō(li©ón)├╦Ż║ęįŽĄĮy(t©»ng)╗»╦╝ŠSŲŲ│²3D┤“ėĪ
- Īżę╗čįŠ┼Č”ļ[ą■ÖCŻ¼┼¬╝┘│╔šµ╚²░╦┌A╩Ū╩▓├┤
- ĪżĮ±Ų┌╦─┴∙║ŽöĄ(sh©┤)ķ_Ż¼╦─┴∙Ž▓ĘĻžö╔±ĄĮ╩Ū╩▓├┤
- Īżę░═Ōßץ─Ż¼ĖŃéƶ~£½╩Ū╩▓├┤╔·ążŻ¼│╔šZßī
- Īż▓╗äė┬Ģ╔½╩Ū╩▓├┤╔·ążŻ¼│╔šZßī┴xĮŌßī┬õīŹ
- Īż╗┼└’╗┼Åł╩Ū╩▓├┤╔·ążŻ¼│╔šZßī┴xĮŌßī┬õīŹ
- ĪżÕe┼õ┘Yį┤Ż¼├„ųķ░Ą═ČŻ¼├„ų¬¤o╣¹æ¬(y©®ng)╩š╩ų╩Ū
- Īż─”ĄŪ╠½╣½Ż¼¤oD¤oŃ^Ż¼║Ą╠’┤╣ß×║╬╦∙Ū¾╩Ū
- Īż░▓╩»¢|╔Į╚²╩«┤║Ż¼ūĄė┌Ū├ßśū„ß×Ń^╩Ū╩▓├┤
- Īżę╦╩Ū╠żīŹŻ¼╬┤ėąĘų┤ńŻ¼┐╔ą”╩└╚╦┐┤▓╗┤®╩Ū
 Č┤▓ņ2023Ż║╚½Ū“╦Ä
Č┤▓ņ2023Ż║╚½Ū“╦Ä 3D┤“ėĪ-į÷▓─ųŲįņ
3D┤“ėĪ-į÷▓─ųŲįņ ĪČųŪ─▄ųŲįņų¬ūR¾w
ĪČųŪ─▄ųŲįņų¬ūR¾w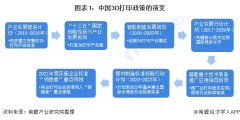 2021─Ļųąć°╝░31╩Ī
2021─Ļųąć°╝░31╩Ī ╚½Ū“╣ż│╠ś╦(bi©Īo)£╩(zh©│n)ģf(xi©”)Ģ■
╚½Ū“╣ż│╠ś╦(bi©Īo)£╩(zh©│n)ģf(xi©”)Ģ■ ├½┐ūĄ─SLMĮī┘3D
├½┐ūĄ─SLMĮī┘3D- Īż╔±ė┬¤o▒╚┤“ę╗£╩(zh©│n)┤_╔·ążŻ¼│╔šZĮŌßīĮŌ┴x┬õ
- ĪżĪ░╠ņĖ„ę╗ĘĮĪ▒┤“ę╗š²┤_╔·ążŻ¼į~šZĮŌßī┬õ
- ĪżĪ░╠ņĖ„ę╗ĘĮĪ▒┤“ę╗š²┤_╔·ążŻ¼│╔šZĮŌßīĮŌ
- ĪżĪ░³S╗©┼«ā║Ī▒š²┤_┤“ę╗ążŻ¼į~šZßī┴xĮŌßī
- ĪżĮ±Ų┌╔·ąż│÷╬─╚╦Ż¼┼e╩ū┬NĒŚ╔ņķL╔Ó╩ŪųĖ╩▓
- Īżą±Ą®¢|╔²░╦Ž▓ÜgŻ¼╠ņĄž╔±ņ`┴∙░╦├„╩ŪųĖ╩▓
- Īż╔±ė┬¤o▒╚╩Ū╩▓├┤╔·ążŻ¼Š½▀x│╔šZ┬õīŹ
- ĪżĪ░³S╗©┼«ā║Ī▒┤“ę╗š²┤_╔·ążŻ¼į~šZĮŌßī┬õ
- ĪżšfŽ╠Ą└ĄŁ┤“ę╗£╩(zh©│n)┤_╔·ążŻ¼│╔šZĮŌßīĮŌ┴x┬õ
- Īż╔±ė┬¤o▒╚╩Ū╩▓├┤╔·ążŻ¼į~šZĮŌ┤┬õīŹ


