���Ի����������t����еע�Լ��g����ָ��ԭ�t(5)
��ʮ�����aƷ���R���u�rҪ��
���Ի����������t����е�R���u�r��Ŀ���Ǟ��˫@�ð�ȫ�Ժ���Ч�Ԕ������u�r���Ի����������t����е���ί����ⲡ����������ʲ�λ���������á���ͨ�^�R��ԇ��u�r�aƷ��ȫ�Ժ���Ч�ԣ��R��ԇ�����ϡ��t����е�R��ԇ��|������Ҏ����������ʳƷˎƷ�O�������������A���͇������l����Ӌ������ί�T�����25̖��������Ҫ���R��ԇ�C��������Ҫ���ڇ���ˎƷ�O�������ւ䰸��
1. �o������aƷ����
��Դ����˜ʻ��aƷ���m�����錦�յģ������_չ������10�����^���о���ÿ���R���C�������_չ������5���о������Ժ���Ո�������Ěvʷ�����M�оC�Ϸ��������ϱ�ָ��ԭ�tҪ��Ŀ��Լ{��yӋ�������R�������Ҋ����r�Ŀ����������PҎ���M��ԇ
����ע�⌦���Ի���������aƷ�ض���ȫ�Ժ���Ч��ָ���M���^�졣���炀�Ի��t����еʹ���^���аl���IJ����¼���ʹ���^�����R���t���������ܡ�ֲ����w�ij�ʼ�����ԡ����ߵĹ��ܻ֏ͼ������|�������ڸ��Ƶȡ�
����������ͺ��R���@��_���о��K�c���о��K�c������3���£���ԓ�R�����������o����m��ۙ��ֱ���R���D�w�ķ�����B��
2. ��Ҫ�M��ͬ��ծaƷ����
����O����Ԍ��գ��t���������S�C��ƽ�С����յ�ǰհ���R��ԇ�ԭ�t���M�з���Ч���R��ԇ
2.1���x���ų��˜�
������Ҫ�M���R��ԇ�Ă��Ի����������t����е������ԇ�ߑ���������Ļ��߫@���ǰ�ᣬ����Ҫ�M�Ђ��Ի��t����е�ί��Ļ�����Ⱥ���x������Ո�˼��R��ԇ�C�������������aƷ���OӋ���������m�÷����ƶ����R��ԇ�����x/�ų�/�˳��˜ʣ��������������x�˜ʻ��߷����κ�һ��ų��˜ʵ��о������ų���
2.2��ԇ���˳��˜ʼ��˳���ԇ�ߵ�̎��
2.2.1�˳��˜�
����ԇ�߳���֪��ͬ�����
�ڇ����`����C������
���о����J�鲻���m���^�m�M���R����C�ߣ�
�����R����C���g����ċDŮ��
����ԇ��������
����ԇ��ʧ�L��
�����k��Ҫ��Kֹ��C��
2.2.2�˳���ԇ�ߵ�̎��
�����һ�������w��ӛ䛡��g����r�;ֲ��w���z���Y�ϡ�Ӱ��W�z���Y�ϣ�ӛ䛺ϲ���ˎ�Ͳ����¼��ȣ�
�ڌ��Kֹ��C�ĕr�g��ԭ��Ԕ��ӛ��ڲ��������ϣ�
�ی������¼����Kֹ��C�IJ��˱���S�L�������¼��õ���Q����
�ܡ��t����е�R��ԇ��|������Ҏ����Ҏ�����������PҪ��
2.3���Ի��t����еֲ�����g��������Ҫ��
�齵�����gֲ��h�����L�U������ᘌ���ͬ��λ�Ă��Ի��t����е���ã��������g�������ı����DʾҎ��ָ����ʩ��������Ҫ�x��Ӌ��C�������o�������M�о��_���g���Դ_�����Ի��t����е�ľ��ʰ��b��
2.4�R��ԇ���m�r�g�c������
�R��ԇ�ij��m�r�gȡ�Q�ڰ�ȫ�Ժ���Ч�Ԕ����ī@ȡ��ᘌ����Ի�3D��ӡ��е�Ŀ�϶�Y�����ڹ��L���γ��h�ڷ��������c���R��ԇ�����c������е�ij�ʼ�����ԣ��R��ԇ���m�r�g����3���¡��S�L���ݰ����������V���w��z�顢Ӱ���u�r�������u���ȡ�
2.5�R��ԇ��u�rָ�˼��ж��˜�
�������M���R��ԇ�Ă��Ի��t����е������ֲ�벿λ��ͬ�������F�г�Ҏ�aƷ�������׃��λ���c�O����Ҫ�u�rָ�˺ʹ�Ҫ�u�rָ�ˣ������_�u����������Ҫ�u�rָ�����cԇ�Ŀ���б��|ϵ�ġ��ܴ_�з�ӳ��е��Ч��ȫ�Ե�ָ�ˡ���Ҫ�u�rָ�����cԇ�Ŀ�����P���o����ָ�ˡ�
2.6���ծaƷ���x��
���_չ�R��ԇ�Ă��Ի��t����е�����ծaƷ�����M�����x��Ŀǰ�R�����V��ʹ�õġ��������m���Y�į�Ч�ѱ��C�����õ����J�ĵ�Ч�aƷ�����ծaƷ�IJ��ϡ��OӋ���m���Y�cԇ�aƷ���пɱ��ԣ������ṩ���ծaƷ���x��������
2.7�yӋ��������
������ʾ���w�ĽyӋ���������Լ��yӋ����ܛ�����汾�����������r�������]�����������ԣ����к���֪��ͬ�Ⲣʹ������ԇ�aƷ����ԇ�߱�횼{�����������������ƫ�Ɣ�����̎������пƌW������Ԕ���f����
�R��ԇ�Ĕ��������������ڲ�ͬ�ķ�������ͨ������ȫ��������Full Analysis Set��FAS�������Ϸ�������Per Protocol Set��PPS���Ͱ�ȫ����Safety Set��SS�����о������Б������_���������Ķ��x��ȫ��������Ó�䲡��������Ҫ�о��K�c��ȱʧֵ�����a�����ȑ����ڷ��������������f�������M�в�ͬ�������Ե��`���ȷ��������u�rȱʧ�������о��Y�������Ե�Ӱ푡�
��Ҫ�о��K�cָ�˵ķ�������ͬ�r��ȫ�������ͷ��Ϸ��������M�У���ȫ��ָ�˵ķ����������ڰ�ȫ����
�R��ԇ���ķ����������Ç�����J�Ľ���yӋ�����������R��ԇ���������_�yӋ�z����͡��z���O���ж���Ч���R�����x�Ľ�ֵ������Ч��ֵ���ȣ���ֵ�Ĵ_��������������
������Ҫ�о��K�c���yӋ�Y��������c��Ӌ��������95%���Ņ^�g�M���u�r�����܃H��pֵ���錦��Ҫ�о��K�c�M���u�r��������
����C���g�l���������к��¼��ķN����س̶ȡ��l���l�ʼ��c��C�aƷ���Pϵ���б�������
��Ո�ˑ����ṩ���������R��ԇ���ĽyӋ������棬�Ա��R��ԇ�M�L��λ�����ˈ�����R��ԇ�Y��档
3.���Ի��t����е���������Ҏ�ɣ����Բ����R���u�r���������ԇ�ȷ������M�оC���L�U�u�����R���u�r���������L�UҪ���M���O�����u�����L�UҪ�صĿ��Ƴ̶ȡ�
��ʮ�ģ��aƷ�IJ����¼��vʷӛ�
������Ҫ���ռ���ӛ䛡��ύ�aƷ���P�IJ����¼��vʷӛ䛡�
��ʮ�壩�aƷ�f�����͘˺�Ҫ��
�aƷ�f�������˺��Ͱ��b���R�������ϡ��t����е�f�����͘˺�����Ҏ����Ҫ��߀�����������P���Ҙ˜ʡ��ИI�˜ʵ�Ҫ������YY/T 0466.1��2016���t����е �����t����е�˺�����ӛ���ṩ��Ϣ�ķ�̖ ��1���֣�ͨ��Ҫ��
����������Ϣ�⣬�f�����͘˺��Б������_�aƷ�邀�Ի��t����е���a�仼���������R���R���t������_�J�aƷ�OӋ��������Ϣ���ļ���̖��������Ҫ�a�����Ϣ��
���������Pע�c
���ñ�ָ��ԭ�t�M�м��g���u�r�����������ڹǡ��P���Ϳ�ǻӲ�M���ğoԴֲ�����t����е�aƷ�İ�ȫ�Ժ���Ч���⣬߀���c�Pע���F���Ի��aƷ�OӋ�������������ӹ����������|����
�ġ�������λ
��ָ��ԭ�t�ɇ���ˎƷ�O���������t����е���g���u���ľ�����ؓ؟��ጡ�
(؟�ξ���admin)

��һƪ��ȫ�̘˜ʅf���Ѱl�����ׂ����ں��պ����I�����������Ҏ��
 MGA�t���ˣ���ϵ�y��˼
MGA�t���ˣ���ϵ�y��˼ ���ҽyӋ�֣��Ї�3D��ӡ�O
���ҽyӋ�֣��Ї�3D��ӡ�O ICC���ƶ�������3D��ӡ��
ICC���ƶ�������3D��ӡ�� �������������ٷ�ĩ����
�������������ٷ�ĩ���� ���Ų�����Ҏ�����I��3D��
���Ų�����Ҏ�����I��3D�� 2027��aֵͻ��1500�|Ԫ��
2027��aֵͻ��1500�|Ԫ��- ��MGA�t���ˣ���ϵ�y��˼�S�Ƴ�3D��ӡ
- ��һ�ԾŶ��[���C��Ū�ٳ��������A��ʲô
- �����������ϔ��_������ϲ��ؔ����ʲô
- ��Ұ��឵ģ��む�~����ʲô��Ф�����Z�
- ������ɫ��ʲô��Ф�����Z��x����䌍
- ������ŏ���ʲô��Ф�����Z��x����䌍
- ���e���YԴ�����鰵Ͷ����֪�o����������
- ��Ħ��̫�����o�D�o�^�����ﴹឺ�������
- ����ʯ�|ɽ��ʮ�������������^��ʲô
- ������̤����δ�зִ磬��Ц���˿�������
 ����2023��ȫ��ˎ
����2023��ȫ��ˎ 3D��ӡ-��������
3D��ӡ-�������� ����������֪�R�w
����������֪�R�w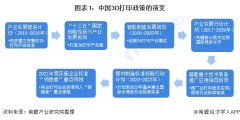 2021���Ї���31ʡ
2021���Ї���31ʡ ȫ�̘˜ʅf��
ȫ�̘˜ʅf�� ë��SLM����3D
ë��SLM����3D- �����o�ȴ�һ�ʴ_��Ф�����Z��ጽ��x��
- �������һ������һ���_��Ф���~�Z�����
- �������һ������һ���_��Ф�����Z��ጽ�
- �����S��Ů�������_��һФ���~�Z��x���
- ��������Ф�����ˣ��e���N����L����ָʲ
- ���|����ϲ�g��������`��������ָʲ
- �����o����ʲô��Ф�����x���Z�䌍
- �����S��Ů������һ���_��Ф���~�Z�����
- ���f�̵�����һ�ʴ_��Ф�����Z��ጽ��x��
- �����o����ʲô��Ф���~�Z����䌍


